







En el complejo, variable y económicamente restrictivo escenario sanitario actual, la utilización de la tecnología es determinante para el monitoreo, prevención, diagnóstico, terapéutica y rehabilitación de la salud humana. El campo de aplicación de los equipos y dispositivos médicos es amplio y evoluciona aceleradamente a través de los avances tecnológicos, convirtiendo este mercado en uno de los más dinámicos y de rápida expansión, especialmente en los países industrializados [1]. Las exigencias para el comercio en este sector se incrementan a nivel internacional buscando altos niveles de calidad relacionados con el desempeño y la seguridad de los productos a fin de garantizar principalmente la seguridad del usuario ya sea paciente, médico u otra parte interesada.
La Organización Mundial de la Salud (OMS), el Foro Internacional de Reguladores de Dispositivos Médicos (IMDRF por sus siglas en inglés) y otras organizaciones internacionales trabajan con la intención de llegar a la armonización global de los requisitos para cumplir con las regulaciones de equipos médicos y facilitar el reconocimiento mutuo de los registros otorgados por las distintas agencias en el mundo. A pesar de ello, las exigencias nacionales y regionales no son todas iguales [2]. Coexisten diversos modelos legislativos que, aunque con similitudes, traen diversas implicaciones para las empresas, especialmente de los países en desarrollo, que son sin dudas las más afectadas por esta multiplicidad. Las diferencias en las regulaciones de los países obliga a los fabricantes y distribuidores a someterse a variados procesos de evaluación y certificación, muchas veces para demostrar mediante distintas y costosas auditorías el cumplimiento de requisitos que son esencialmente iguales.
En el caso de la Unión Europea (UE), las exigencias son muy altas en cuanto a la evaluación del cumplimiento de los requisitos esenciales de seguridad y eficacia de los equipos médicos [3]. Estos requisitos forman parte de complejos marcos regulatorios recogidos en sus directivas comunitarias para los cuales la demostración de la conformidad está basada, fundamentalmente, en el cumplimiento de normas europeas armonizadas. Los procedimientos de evaluación de la conformidad son el trámite necesario para la colocación del marcado CE en los productos, lo cual permite su libre circulación dentro del mercado de la Unión Europea. Sin embargo, algunos consideran estas exigencias como barreras de entrada por lo que conviene analizar si efectivamente lo son, pues muchas veces se utiliza este concepto de forma errónea [4].
Según la Organización Mundial del Comercio (OMC), el objetivo del Acuerdo sobre Obstáculos Técnicos al Comercio (Acuerdo OTC) es que los reglamentos técnicos, las normas y los procedimientos de evaluación de la conformidad no sean discriminatorios ni creen obstáculos innecesarios al comercio. Al mismo tiempo, el acuerdo reconoce el derecho de los Miembros de la OMC a aplicar medidas para alcanzar objetivos normativos legítimos, tales como la protección de la salud y la seguridad de las personas o la protección del medio ambiente. Para ello se recomienda firmemente a los Miembros que basen sus medidas en normas internacionales como medio de facilitar el comercio y crear un entorno comercial previsible [5].
Este trabajo pretende analizar si las exigencias respecto al requisito de seguridad del producto y del marcado CE de conformidad europea son posibles barreras técnicas de entrada al mercado europeo de equipos médicos y su percepción por parte de los fabricantes. Esta valoración forma parte de una investigación mucho más amplia, actualmente en ejecución.
Respecto al tema de las barreras técnicas en los equipos médicos existe poca investigación empírica y es dispersa la bibliografía. La mayor parte de la información colectada en este trabajo se obtuvo a partir de una revisión de la literatura en bases de datos indexadas como la WOS y Science Direct; también de repositorios de organizaciones de alcance mundial como OMS y OMC.
Asimismo, se ha preparado un estudio de casos siguiendo la metodología de Yin (1984) con un grupo de empresas del sector, empleándose un cuestionario adaptado de una investigación previa realizada en Brazil, así como entrevistas semi-estructuradas a personal de estas entidades. Todo ello permitirá generar hipótesis sobre la manifestación de esta barrera en la práctica y las medidas sobre cómo superarlas, con lo cual se completará la investigación en curso.
Las barreras comerciales se emplean como mecanismos para restringir el acceso de los productos importados en el comercio internacional. Se utilizan tanto barreras arancelarias como no arancelarias. Estas últimas, incluyen las barreras técnicas al comercio derivadas del uso de normas, reglamentos técnicos o procedimientos de evaluación de la conformidad; considerada una de las principales dificultades que los exportadores encuentran para insertar sus productos en mercados externos. Los procedimientos de evaluación de la conformidad son procedimientos técnicos, de prueba, verificación, inspección o certificación, que sirven para determinar que los productos cumplen las prescripciones establecidas en los reglamentos y las normas. Estos procedimientos deben ser transparentes y aplicarse de manera no discriminatoria para evitar que se conviertan en instrumentos proteccionistas y en un obstáculo técnico al comercio si se emplean de forma ilegitima [6]. En las últimas décadas, el número de normas y reglamentos técnicos adoptados por los países ha aumentado considerablemente. Esta intensificación de la política de reglamentación se atribuye a mejoras en el nivel de vida global, lo que ha impulsado la demanda de productos seguros y de alta calidad por parte de los consumidores [7]. Particularmente en el sector sanitario esta tendencia es notable, tanto en Europa como fuera del continente [8, 9].
A nivel mundial, las agencias sanitarias establecen programas regulatorios con requisitos que varían en dependencia de la clase de riesgo del equipo médico, tomando en cuenta la vulnerabilidad del cuerpo humano ante un potencial fallo o mal funcionamiento del producto. La autorización de la comercialización de un equipo o dispositivo médico implica que se han cumplido los requisitos esenciales de seguridad y eficacia, características que deben ser evaluadas en la aplicación específica en que se pretende utilizar el producto. El cumplimiento de los requisitos esenciales es obligatorio y debe garantizarse desde la etapa de diseño, durante la fabricación e incluso en la posventa, especialmente para los equipos médicos de medio y alto riesgo. Estos se reflejan en una serie de requerimientos particulares, los cuales son evaluados por las agencias regulatorias sobre la base de la información brindada por el fabricante y considerando los estándares y regulaciones armonizadas a nivel internacional, entre las que destacan la familia de normas ISO 13485 para los sistemas de gestión de calidad, la ISO 14971 para la gestión del riesgo de los equipos médicos y las IEC 60601-1 de Seguridad eléctrica.
Exigencias técnicas en la Unión Europea. Procedimientos para la evaluación de la conformidad europea (Marcado CE) y requisitos de seguridad del producto.
Dada la diversidad tecnológica y capacidad de innovación en el campo de los equipos médicos que existe en la Unión Europea los requerimientos son elevados. En esta región, el 95 % de las empresas dedicadas a las tecnologías sanitarias son PYMES y este sector es uno de los que genera mayor cantidad de empleo con ventas totales que superan los 100 billones de euros [10]. De ahí, que sea un campo fuertemente regulado y de relevancia internacional.
A diferencia de Norteamérica, en Europa las Directivas de productos sanitarios son aprobadas por el Parlamento Europeo y luego se transponen o adaptan en cada país miembro de la comunidad, lo cual permite que los países europeos incluyan requisitos nacionales específicos. Esta legislación establece los requisitos reglamentarios de obligatorio cumplimiento, las bases normativas para la evaluación premercado así como la existencia de Organismos Notificados que de manera descentralizada otorgan la marca CE siguiendo procedimientos de evaluación determinados [11]. Un aspecto que puede ser favorable ha sido la reciente revisión de las disposiciones europeas con el objetivo de evitar divergencias entre las transposiciones en los diferentes países (ver figura 1).
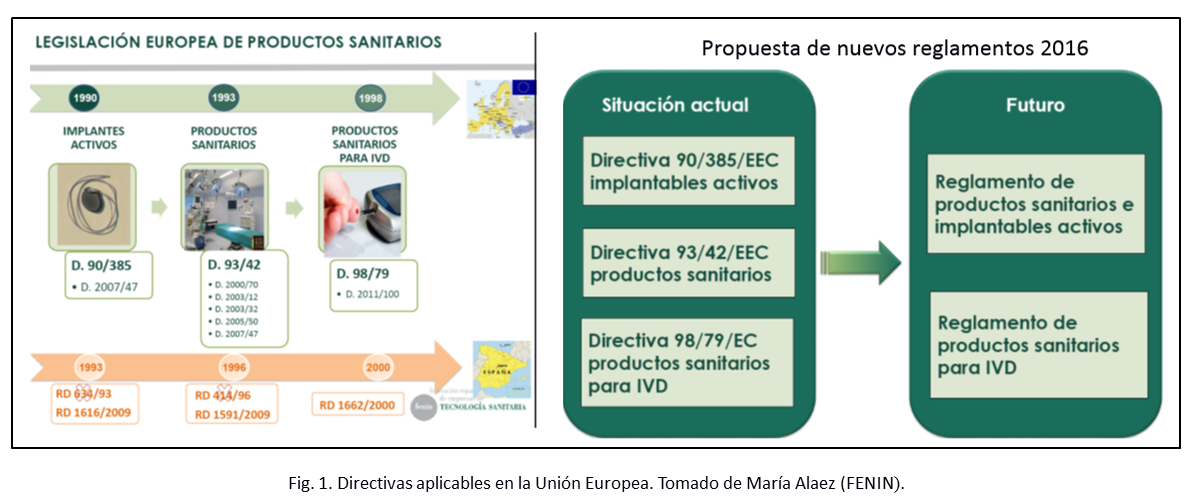
Para poder comercializar un producto en el mercado europeo es necesario cumplir con las directivas específicas que se establecen de acuerdo al tipo de equipo médico que se trate. Cada una de ellas contiene requisitos del sistema de gestión de la calidad (SGC) del fabricante y requisitos del producto en cuanto a seguridad y eficacia. En cualquier caso, se puede presumir conformidad del cumplimiento con los requisitos esenciales establecidos en la directiva cumpliendo con normas europeas armonizadas, si éstas no existen se recomienda la aplicación de normas internacionales y por último la aplicación de normas nacionales. El establecimiento de normas europeas con muy altos niveles de exigencia es una fuente potencial de dificultades técnicas, pues restringe el acceso de los exportadores con menor capacidad tecnológica, afectando principalmente a los productores de los países en desarrollo [12]. Pero vale señalar que este tipo de restricción comercial no es necesariamente negativa, ya que produce beneficios para los consumidores (europeos y del resto del mundo) en términos de productos de mejor calidad.
Una vez que el fabricante, sea europeo o no, conoce la directiva aplicable, este puede seguir diferentes rutas para lograr la conformidad con la misma (ver fig. 2) presentando una documentación sobre el diseño, los procesos de fabricación y esterilización, las pruebas de funcionamiento, los ensayos clínicos, los materiales de envasado, las normas técnicas que cumplen y la información que acompaña al producto. Los procedimientos varían desde una autodeclaración del fabricante (para los de menor riesgo o clase I) hasta una evaluación completa por un organismo notificado que puede incluir una auditoría en las instalaciones donde se fabrica el producto. Si el resultado de las comprobaciones es favorable, se emite un certificado de conformidad que permite colocar el número de organismo notificado junto con el distintivo CE en el producto, lo que indica que cumple con los requisitos de la reglamentación [13]. Obtenido este marcado puede comercializarse el producto sanitario en todos los países de la UE sin necesidad de nuevas evaluaciones, aunque el fabricante debe comunicar a las autoridades sanitarias de su primera puesta en el mercado para lo cual debe pagar importantes tasas.
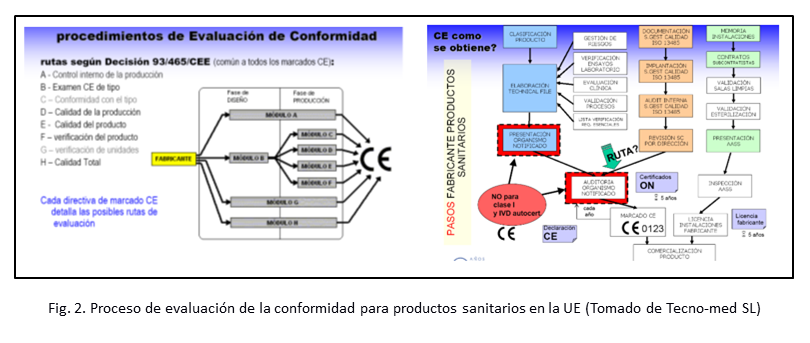
Obtener el marcado CE significa el compromiso del fabricante con 4 aspectos fundamentales: garantía de información (documentación técnica completa), garantía de calidad (sistema de calidad conforme a normas armonizadas), garantía de funcionalidad (evaluación del usuario, ensayos clínicos, evaluación del funcionamiento, validación del producto) y garantía de seguridad (ensayos según normas armonizadas, verificación del producto) [14].
Para garantizar el requisito esencial de la seguridad, específicamente en los equipos electromédicos, es vital demostrar el cumplimiento con las normas técnicas que han sido adoptadas como referencia en la UE como la serie 60601 de la IEC (siglas en inglés de Comisión Electrotécnica Internacional). Estas normativas generales, particulares y colaterales establecen requisitos de producto y de procesos para asegurar el desempeño básico del equipamiento de modo que no afecte la seguridad del paciente, del operador o del medio ambiente.
Comprobar el cumplimiento de estas especificaciones en los equipo médicos que se exportan hacia la UE significa realizar un conjunto de procesos (análisis, pruebas, ensayos) en laboratorios acreditados por las autoridades sanitarias correspondientes, lo cual implica determinados desafíos técnicos y económicos a las empresas que buscan introducir sus productos en el mercado europeo. Cuando la legislación europea impone la necesidad de recurrir a procedimientos de evaluación de la conformidad de terceras partes, la aprobación del producto final sólo puede ser concedida por ciertas organizaciones en la región designada por los países miembros. Por lo tanto, los productores extranjeros están obligados a incurrir en costos duplicados y pruebas de certificación de sus productos para ser aprobados.
Además, el sistema europeo de evaluación de la conformidad también tiene consecuencias adversas sobre la competitividad de los productores foráneos lo que ha obstaculizado muchas veces las exportaciones de otros países hacia la UE. El concepto de enfoque global de evaluación de la conformidad se estableció en la región con el fin de reducir costos y aumentar el grado de confianza en tales procedimientos, consolidando el principio de reconocimiento mutuo de normativas y regulaciones para optimizar el comercio interno. Sin embargo, se considera que existen factores positivos y negativos en esta cuestión. Por un lado, al unificarse las normas y procedimientos adoptados en varios países europeos se disminuyen los costos asociados a la obtención de información y adaptación a los criterios de cada país. Pero al mismo tiempo, obliga a las empresas foráneas a gastar más, toda vez que deben probar sus productos conforme a estándares puramente europeos del CEN, CENELEC y ETSI[1], sin lo cual no se consideran aptos para su comercialización en la región. Esto deriva en una posible barrera técnica al comercio ya que las empresas de fuera de la Unión Europea no participan en el desarrollo de dichos estándares, estableciéndose un sesgo en ese proceso en beneficio de las empresas de la región y generando mayores dificultades para las empresas extranjeras [15].
Con el objetivo de conocer la percepción de los fabricantes respecto al tema de las barreras al comercio en la UE e identificar su existencia e impacto, se ha diseñado un estudio de casos exploratorio con un grupo de empresas del sector de equipos médicos, europeas y extra-europeas. Estas empresas cuentan con alguna experiencia exportadora y son de diversos tipos (pequeñas y medianas en su mayoría).
Para ello se adaptó el cuestionario desarrollado por Grecco-Moraes en 2007, contentivo de cinco dimensiones que agrupan las principales variables de interés relacionadas con la exportación de equipos médicos hacia Europa [16]. Estas dimensiones comprenden la caracterización de las empresas y sus productos; la comparación de exigencias entre el mercado europeo y el externo en cuanto a reglamentación y normas aplicables (de calidad, de gestión de riesgos y de seguridad); la identificación y ponderación del grado de importancia que le dan los fabricantes a las dificultades encontradas durante sus exportaciones; el nivel de inversiones en I+D+i que realizan las empresas así como indagarles sobre los factores que consideran determinantes para superar estas barreras. Este estudio está actualmente en ejecución y sus resultados se publicarán próximamente.
[1]Comité Europeo de Normalización (CEN), Comité Europeo de Normalización Electrotécnica (CENELEC), European Telecommunications Standards Institute (ETSI) o Instituto Europeo de Normas de Telecomunicaciones (www.cen.eu)
Por lo visto, las exigencias europeas para la evaluación de la conformidad de los equipos médicos y la obtención del marcado CE son consideradas en su mayoría como barreras de entrada no arancelarias de carácter técnico. Ya sean exigencias obligatorias (como las directivas y reglamentos) o voluntarias como las normas, se considera que estas se utilizan como formas disimuladas de protección de los mercados nacionales restringiendo el acceso a fabricantes extranjeros que deben adecuar sus productos y procesos a rigurosos procedimientos para poder comercializarlos dentro del mercado europeo. No obstante, algunos autores señalan que existe cierta incomprensión del concepto de barreras técnicas ya que muchas veces estas se asocian a las propias dificultades que tienen los exportadores, en el orden técnico, financiero y/o cultural, para cumplir los requerimientos establecidos; especialmente cuando se trata de pequeñas y medianas empresas (Pymes) de menor capacidad tecnológica [17].
Estudios realizados en empresas del sector de mercados emergentes como la India reconocen que las certificaciones como el marcado CE son una importante alternativa, al mismo tiempo que afirman que el alto costo de tales procedimientos y/o la lejanía de estas agencias u organismos notificados son serios obstáculos y dan lugar a retrasos para la comercialización [18]. En tanto, otro enfoque se encuentra en estudios llevados a cabo en los cuatro países europeos con los mayores gastos en dispositivos médicos (Alemania, Francia, Italia y Reino Unido), el cual revela sus políticas para encontrar el correcto equilibrio entre mejorar el acceso a nuevos productos y restringir las fuerzas del mercado a fin de contener costos y garantizar asequibilidad [19].
Asimismo, algunos países establecen requisitos regulatorios adicionales para los productos sanitarios. En España, por ejemplo, el Real Decreto 1591/99 obliga al exportador no solo a la evaluación y registro del equipo médico de conformidad con las normativas vigentes sino también a la obtención de la licencia sanitaria del fabricante, importador y distribuidor ante la autoridad competente, para lo cual debe cumplir múltiples, complejos y en no pocas ocasiones costosos requerimientos.
En general, superar los obstáculos técnicos al comercio conlleva elevados costes tanto iniciales como continuos [20]. La propia OMC ha reconocido que entraña costos considerables para los productores y los exportadores, aunque le resulta difícil hacer una estimación precisa de la repercusión en el comercio internacional de la necesidad de cumplir distintas normas y reglamentos técnicos extranjeros. Señala que estos costos están relacionados con la información correspondiente a la evaluación y traducción de reglamentos extranjeros (en algunos casos divergentes), la contratación y formación de expertos técnicos que expliquen dichos reglamentos y la divulgación sobre los productos. También, costos por pérdidas de economía de escala debido a la adaptación de las instalaciones de producción para que se ajusten a las prescripciones extranjeras de diferentes mercados, muchas veces diversas. Igualmente, declara que se incurre en costos de evaluación de la conformidad ya que su confirmación requiere pruebas, certificaciones o inspecciones efectuadas por laboratorios u órganos de certificación, normalmente a expensas de la empresa; y otros costos imprevistos u ocultos que ponen en desventaja al exportador respecto a las empresas nacionales [21].
Los elevados costos pueden desalentar a los fabricantes de tratar de vender en el extranjero, además de que limita a las Pymes en su inserción al comercio internacional. Al no haber disciplinas internacionales, se corre el riesgo de que se adopten y apliquen normas y reglamentos técnicos con el único objeto de proteger a las ramas de producción nacionales. En el Acuerdo OTC se insta a los Miembros de la OMC a entablar negociaciones con otros Miembros para la aceptación mutua de los resultados de sus respectivos procedimientos de evaluación de la conformidad, aunque sean diferentes. Así, el producto solo se probaría en el país de origen y los resultados se aceptarían en el resto de los mercados evitando duplicidad.
La existencia de un alto grado de confianza en los organismos y laboratorios de prueba constituye una condición previa para el buen funcionamiento de un acuerdo de reconocimiento mutuo. Lo cual puede contribuir a evitar el costo de la multiplicidad, muchas veces provocado porque los expertos encargados de las pruebas no están de acuerdo con procedimientos de prueba óptimos, por inercia burocrática o incluso por manipulación del proceso de prueba por grupos proteccionistas. Sea cual fuere la razón, esa heterogeneidad de procedimientos y métodos aumenta considerablemente el costo para los productores que venden en diversos mercados [21].
En la práctica, ya se ha demostrado que es posible realizar estos acuerdos. De hecho, se manejan dentro de los estados miembros de la UE e incluso se han suscrito entre esta y la Administración de Alimentos y Medicamentos de Estados Unidos[1] (FDA por sus siglas en inglés). Siguiendo la intención de armonización cada vez son más los países que están adoptando dentro de sus sistemas regulatorios nacionales las aprobaciones de otros países, especialmente de los líderes del sector. Recientemente 5 países euroasiáticos adoptaron acuerdos similares entre sí [22].
Entonces, valdría considerar la adopción de mecanismos de reconocimiento mutuo en los casos de países que cuenten con una infraestructura regulatoria sólida y cualificada. Extender estos acuerdos hacia otras regiones o bloques fuera de la Unión Europea (como el MERCOSUR de América Latina u otros) abriría las puertas de acceso a este importante mercado, permitiendo que productos innovadores y debidamente registrados en otros países puedan ser comercializados en países europeos, sin que sus fabricantes y/o exportadores tengan que sufrir la multiplicidad de ensayos, certificaciones, inspecciones y altos costos que actualmente genera el proceso para la obtención del marcado CE.
Por otro lado, los países de fuera de la UE podrían influenciar indirectamente el proceso de normalización europeo, participando en los comités técnicos de la ISO (Organización Internacional de Normalización) y de la IEC, ya que los comités europeos CEN y CENELEC se han comprometido a no promulgar normas relativas a productos para los que ya existan o estén en estudio normativas de estas organizaciones internacionales [14]. Así, se podría encontrar una vía para evitar el surgimiento de nuevas barreras de entrada al mercado europeo. Aunque de igual forma ya se trabaja desde la propia Comisión Europea en la prevención de la creación de barreras técnicas al comercio, para lo cual ha implementado algunos procedimientos generales [23].
[1] Food and Drug Administration (FDA). Sección de Equipos Médicos. Disponible en: http://www.fda.gov/cdrh.
La seguridad de los productos sanitarios es imprescindible como requisito esencial y se prioriza en función de la seguridad de los usuarios. El procedimiento de evaluación de la conformidad y marcado CE, son paradigmas de actuación que se centran en el uso de normas europeas armonizadas cuyo contenido técnico es compatible con las prácticas internacionales y devienen en un instrumento útil tanto para los fabricantes como para los organismos evaluadores. Amén de su importancia y legitimidad, ambos aspectos actúan como freno para la entrada de equipos médicos a la UE por cuanto resultan muy costosos para las empresas que deben multiplicar esfuerzos para satisfacer las exigencias de este riguroso mercado.
Si bien es cierto que los programas regulatorios para los productos sanitarios de la UE fomentan la igualdad de condiciones técnicas entre los fabricantes, muchos lo aprecian como barreras o trabas para acceder al mercado sin considerar que con estas medidas se fija una cota mínima de calidad, seguridad y eficacia en los productos comercializados, lo que contribuye a que sean más competitivos y beneficia al propio desarrollo del sector. No obstante, es innegable que estos requisitos reglamentarios suponen una carga económica adicional sobre las empresas, especialmente Pymes y aquellas de países en desarrollo.
La legislación europea de dispositivos médicos es una regulación moderna que ha sido objeto de modificaciones y ampliaciones que la han consolidado a fin de lograr un marco regulatorio más adecuado, transparente y sostenible. Este año la UE aprobó los borradores de los nuevos reglamentos para los productos sanitarios que amplía el campo de aplicación a nuevos productos e introduce varios cambios relevantes. Novedad que tiene un impacto importante no solo en los productores y distribuidores, para los que se presume un incremento en tiempo y costes, sino también en el resto de los agentes implicados (pacientes, profesionales sanitarios, reguladores, organismos notificados, laboratorios de referencia) toda vez que se incrementan las exigencias a todos los niveles.
La intención de llegar a la armonización global de los requisitos para los equipos médicos y al reconocimiento mutuo internacional sigue siendo una meta tanto para la OMS, el actual IMDRF y todos los actores y partes interesadas imbricadas en este complejo y pujante sector.
Los autores agradecen el apoyo ofrecido por la Agencia Española de Cooperación Internacional (AECID), la Asociación Universitaria Iberoamericana de Posgrado (AUIP) y la Universidad de Málaga para la realización del proyecto que contempla esta investigación.
- Maresova, P., Penhaker, M., Selamat, A., & Kuca, K. (2015). The potential of medical device industry in technological and economical context. Therapeutics and clinical risk management, 11, 1505.
- World Health Organizations (2011). First WHO Global Forum on Medical Devices: control, outcomes and future actions (disponible en: www.who.int/medical_devices).
- Chai, J. (2000). Regulation of medical devices in the European Union. Journal of Legal Medicine, 21(4), 537-556.
- Carlton, D. W. (2004). Why barriers to entry are barriers to understanding (No. w10577). National Bureau of Economic Research.
- Organización Mundial del Comercio (WTO). https://www.wto.org/spanish/tratop_s/tbt_s/tbt_s.htm (consultado, Mayo 2015)
- Sistema de Información sobre Comercio Exterior de la OEA. http://www.sice.oas.org/dictionary/TBT_s.asp (consultado, Marzo 2016).
- Ministerio de Industria y Comercio de la República Dominicana (2011). Publicación de la secretaria de comercio exterior “Libre comercio”, en: http://www.mic.gob.do/media/64872/barreras%20tecnicas%20al%20comercio.pdf
- Altenstetter, C. (2003). EU and member state medical devices regulation. International journal of technology assessment in health care, 19(01), 228-248.
- Lamph, S. (2012). Regulation of medical devices outside the European Union. Journal of the Royal Society of Medicine, 105(suppl 1), S12-S21.
- European Commission (EC). En: http://ec.europa.eu/growth/sectors/medical-devices/index_en.htm (consultado Mayo 2016).
- French-Mowat, E., & Burnett, J. (2012). How are medical devices regulated in the European Union?.Journal of the Royal Society of Medicine, 105(suppl 1), S22-S28. (describe proceso del marcado CE)
- Malkin, R. A. (2007). Barriers for medical devices for the developing world. Expert review of medical devices, 4(6), 759-763.
- Agencia Española de Medicamentos y Productos sanitarios (AEMPS). 2009. Memoria de actividades. En: www.aemps.es.
- Consultoría Tecno-med Ingenieros. 2009. Curso sobre distribución y servicio de asistencia técnica. En: www.tecno-med.es
- Silva Ruiz, M., Brescansin, A., Coelho Cerântola, A. P., Lanari Ozolins, A., de Oliveira e Aguiar, A., Casteli Figueiredo Gallardo, A. L., ... & Ytoshi Shibao, F. (2014). O setor de eletroeletrônicos: aspectos técnicos, econômicos, regulatórios e ambientais.
- D'elia, M. A. G., & Zouain, D. M. (2008). Superação das barreiras técnicas ao comércio internacional pelas pequenas e médias empresas de base tecnológica-a exportação de produtos eletromédicos para a união européia. Revista de administração e inovação-rai, 5(1).
- Garrido, A. E. (2004). As barreiras técnicas ao comércio internacional. Instituto Nacional de Metrologia, Normalização e Qualidade Industrial, Rio de Janeiro. Access in March, 31, 2005.
- Jarosławski, S., & Saberwal, G. (2013). Case studies of innovative medical device companies from India: barriers and enablers to development. BMC health services research, 13(1), 1.
- Schreyögg, J., Bäumler, M., & Busse, R. (2009). Balancing adoption and affordability of medical devices in Europe. Health Policy, 92(2), 218-224.
- Togan, S., & Dogan, S. (2009). La eliminación de obstáculos técnicos al comercio en el contexto de la unión aduanera entre Turquía y la Unión Europea. Información Comercial Española, ICE: Revista de economía, (846), 47-64.
- Organización Mundial del Comercio (WTO). En: https://www.wto.org/spanish/tratop_s/tbt_s/tbt_info_s.htm (consultado Abril 2016)
- Grupo Emergo. En http://www.emergogroup.com/es/node/5323). (consultado en Mayo 2016).
- European Commission (EC). En:http://ec.europa.eu/growth/single-market/barriers-to-trade/index_en.htm (consultado en Mayo 2016).
Papers relacionados









