




Fernández Rodríguez, Pablo; Fernández Vilas, Eva; García García, Laura Departamento de Prevención Técnica / Instituto Nacional de Silicosis / Doctor Bellmunt s/n / 33006 Oviedo, España
+34 985 10 78 40 / pfernandez@ins.es
ABSTRACT
ABSTRACT
En el presente trabajo se pusieron a punto metodologías analíticas para la determinación del contenido en sílice presente en la atmósfera de trabajo de explotaciones a cielo abierto, directamente sobre el propio filtro de muestreo (DOF), tanto mediante IR como mediante DRX. Las nuevas metodologías desarrolladas se compararon con los métodos analíticos indirectos empleados en el Laboratorio del Departamento Técnico del INS, actualmente acreditado por ENAC. Las incertidumbres obtenidas con el empleo de los métodos directos fueron un 20% mejores, en términos relativos, debido a la ausencia de etapas de preparación previas al análisis de la muestra.
Palabras clave
Palabras clave
Sílice, explotaciones a cielo abierto, FTIR, DRX
DETERMINACIÓN DE SÍLICE LIBRE CRISTALINA DIRECTAMENTE SOBRE MEMBRANA EN MATERIA PARTICULADA (FRACCIÓN RESPIRABLE)
Introducción
Introducción
En estos últimos años existe un interés creciente por estudiar la exposición de los trabajadores al polvo existente en los lugares de trabajo, especialmente debido al mayor conocimiento acerca de los efectos potenciales sobre la salud de la sílice cristalina presente en un ambiente pulvígeno. La “International Agency for Research on Cancer” (IARC) concluyó en 1997 que la exposición ocupacional a sílice cristalina puede causar cáncer de pulmón en humanos. Más recientemente, el “Scientific Committee on Occupational Exposure Limits” (SCOEL) reconoció que el principal efecto de la sílice cristalina sobre los humanos es la silicosis y que los silicóticos tienen un riesgo más elevado de desarrollar un cáncer de pulmón. Los expertos concluyeron que, dado que no se puede identificar un límite claro a partir del cual comienza a desarrollarse la silicosis, la reducción de la exposición reduciría el riesgo de desarrollar la enfermedad, recomendando un “umbral” o límite de exposición ocupacional por debajo de 0,05 mg/m3. Actualmente, dicho límite todavía no se ha aceptado legalmente a nivel europeo, por lo que son los estadosmiembros quienes, a título individual, están estableciendo límites cada vez más estrictos en respuesta a las recomendaciones IARC y SCOEL.
En España, la Instrucción Técnica complementaria 2.0.02 “Protección de los trabajadores contra el polvo, en relación con la silicosis, en las industrias extractivas” del Reglamento General de Normas Básicas de Seguridad Minera, recientemente aprobada, ha supuesto una modificación del valor límite de 0,25 a 0,1 mg/m3, límite que ya era aplicado en otro tipo de industrias como las del sector químico. Las estadísticas publicadas anualmente por el Instituto Nacional de Silicosis acerca de los nuevos casos de silicosis detectados en nuestro país revelan que la enfermedad no está extinguida. La prevención sigue siendo un factorimportante para evitar el desarrollo de la misma, especialmente cuando estudios médico técnicos llevados a cabo en industrias extractivas del sector del granito, han revelado la existencia de trabajadores que han comenzado a desarrollar la enfermedad con períodos de exposición a la sílice más bajos de los que cabría esperar para el desarrollo de la enfermedad.
La aplicación de límites de exposición ocupacional cada vez más estrictos exige una mayor exactitud y precisión en la determinación de los niveles de sílice, especialmente cuando éstos son bajos. En estos casos la variabilidad de las medidas es elevada y, si no existen los controles apropiados, es probable que la incertidumbre sea mayor de lo permitido en especial si la contribución del muestreo es tenida en consideración, tal y como establece el Comité Europeo de Normalización (CEN) en los requerimientos para el cálculo de la incertidumbre en medidas de higiene ocupacional.
La mayor parte de los análisis de muestras de polvo respirable tomadas en explotaciones mineras a cielo abierto en España son realizados en el Laboratorio del Departamento Técnico del Instituto Nacional de Silicosis. Dependiendo de la matriz en la que se encuentra presente la sílice, su determinación se lleva a cabo mediante Espectroscopía de Infrarrojo con transformada de Fourier (FTIR) o mediante Difracción de rayos X (DRX), en ambos casos empleando métodos indirectos de análisis. En este tipo de métodos el polvo recogido sobre un filtro se recupera y redeposita sobre otro sustrato (una pastilla de KBr en FTIR o una membrana de Ag en DRX) para ser posteriormente analizado. Los métodos indirectos ofrecen la posibilidad de un tratamiento previo de la muestra con objeto de eliminar interferencias. Sin embargo, este proceso adicional incrementa la posibilidad de pérdidas de muestra y bajas recuperaciones, así como los costes del análisis.
En los últimos años algunos organizadores de ensayos de intercomparación han publicado los resultados obtenidos por los laboratorios participantes, llegando a la conclusión de que aquellos que emplean métodos directos de análisis, es decir, métodos en los que el propio soporte de muestreo se introduce en el equipo para su análisis, tienen en general mejores prestaciones analíticas que los laboratorios que emplean métodos indirectos.
En el presente trabajo se ponen a punto metodologías analíticas para la determinación directa de sílice sobre el propio filtro de muestreo, tanto mediante FTIR como mediante DRX, describiendo las cámaras de generación de polvo empleadas para la preparación de los estándares de calibración, el tipo de filtro más adecuado para este tipo de análisis y, en el caso de DRX, comparando dos tipos de detectores. Las nuevas metodologías desarrolladas se comparan con los métodos analíticos empleados actualmente en el Laboratorio del Departamento Técnico del INS, acreditados por la Entidad Nacional de Acreditación (ENAC). Por último se presentan las conclusiones generales del trabajo.
Determinación de sílice libre cristalina mediante métodos directos de análisis
Los métodos directos e indirectos para la medida de la concentración ambiental de un contaminante en atmósferas de trabajo compiten frecuentemente dentro del campo de la higiene industrial. En el caso concreto del análisis de sílice libre cristalina los métodos directos de análisis reducen la manipulación de la muestra al mínimo pero, como el depósito de polvo sobre el filtro de muestreo no es uniforme, las condiciones analíticas deben ser rigurosamente definidas. En particular, los patrones empleados para construir la curva de calibrado deben serobtenidos en las mismas condiciones que las muestras a analizar, y si las operaciones de muestreo y los análisis no son llevados a cabo por el propio laboratorio es importante la existencia de una comunicación fluida entre ambas partes para asegurar una analítica adecuada. Los métodos indirectos, por el contrario, no están sujetos a esta limitación y tienen la ventaja de permitir un tratamiento de la muestra que limite el riesgo de interferencias. Sin embargo, la metodología debe ser estrictamente controlada para minimizar las pérdidas de muestra durante la preparación de la misma.
Se han descrito en la literatura diversas comparaciones de técnicas para el análisis de sílice cristalina en muestras ambientales. Así, el “National Institute for Occupational Safety and Health” (NIOSH) [1] evaluó las prestaciones de dos métodos analíticos mediante DRX e IR a partir de los resultados de ensayos de intercomparación. Los laboratorios participantes recibieron 8 filtros cargados con cuarzo asociado a diferentes matrices, tales como calcita, caolín, talco, óxido de hierro y carbón. Para cada método el error relativo medido se situó en torno al 20%, con similares contribuciones de los errores intra e interlaboratorio. Pickard y colaboradores [2] encontraron resultados similares en el análisis directo de muestras tomadas en diferentes sectores industriales. Lorberau y col. [3] demostraron la existencia de un grado de concordancia entre los resultados obtenidos mediante DRX tanto en análisis directo como en indirecto, si bien las muestras contenían altas proporciones de cuarzo, limitando así la presencia de interferencias. Más recientemente, 30 muestras tomadas en minas de carbón fueron analizadas en 5 laboratorios europeos empleando IR o DRX [4]. Los resultados sugieren que la DRX está menos afectada por el tamaño de partícula y es menos sensible a interferencias potenciales. En los últimos años los proveedores de ensayos de intercomparación han proporcionado información adicional acerca de los resultados obtenidos por laboratorios de higiene industrial que realizan este tipo de análisis. En el caso concreto del “Workplace Analysis Scheme for Proficiency” (WASP) no se encontraron diferencias significativas entre los laboratorios que empleaban IR o DRX mediante análisis directo de los filtros, pero sí con respecto a aquellos que empleaban métodos indirectos [5].
Cámaras para la generación de estándares de calibración
Como ya se ha comentado en la sección anterior, un aspecto importante cuando se pretende emplear métodos directos de análisis consiste en que los estándares de calibración deben ser preparados en las mismas condiciones que las muestras a analizar. El propio filtro empleado en la toma de muestra será el soporte utilizado en IR o DRX para el análisis de la misma. Es por tanto imprescindible disponer de un generador de nubes de polvo para la producción de atmósferas que contengan el estándar de cuarzo a partir del cual se van a preparar los patrones de calibración.
Un esquema del primer prototipo (Cámara A) diseñado en nuestro laboratorio se presenta en la Figura 1. Consiste básicamente en un diseño de vidrio de borosilicato (4) en cuyo interior, en su parte superior, se coloca el muestreador de fracción respirable. El diseño acaba en forma cónica y es aquí donde se deposita el polvo de cuarzo empleado como estándar (en nuestro caso NIST SRM 1878a). Para poner en suspensión el polvo se introduce aire comprimido a través de un tubo de vidrio de 1 mm de diámetro interno (3) conectado por medio de un tubo de tygon a una válvula (1) de volumen ajustable. Esta válvula acumula el aire comprimido procedente de una botella externa y mediante un dispositivo electrónico (2) lo expulsa a intervalos de tiempo regulables entre 1 y 5 segundos. El volumen de vidrio (4) se encuentra cerrado en su parte superior por una tapa de plástico con dos orificios: por uno de ellos se introduce el tubo interno (3) y por elotro se introduce el tubo de plástico que conecta el ciclón o selector de fracción respirable con la bomba de muestreo.
El modo de operación típico con este diseño consiste en introducir en torno a 100 mg de patrón de cuarzo en la base del dispositivo (4). La válvula (1) se regula de modo que sobre el polvo depositado se inyecte un volumen de aire de 10 mL a una presión de aproximadamente 300 psi, a intervalos de 2 segundos durante 1 minuto. Transcurrido este tiempo se detiene la inyección de aire y se pone a funcionar la bomba de muestreo durante el tiempo suficiente para la preparación de estándares, típicamente entre 5 segundos y 2 minutos.
Figura 1.- Cámara A empleada para la preparación de estándares de calibración: 1) Válvula de volumen ajustable; 2) Dispositivo electrónico para controlar la frecuencia de disparo; 3) Tubo interior por donde se introduce el aire comprimido; 4) Dispositivo de vidrio en cuya parte superior se coloca el muestreador y en su base el polvo.

La principal limitación de este diseño desde el punto de vista operativo la constituye el hecho de poder tomar una sola muestra de cada vez, al no poder introducir más de un muestreador en su interior. Por otro lado, el hecho de emplear sucesivos “disparos” de aire en cada toma unido al pequeño volumen del diseño de vidrio (4) provoca que el cuarzo se vaya apelmazando en las paredes, lo que obliga a acondicionar de nuevo el polvo en su base cada 3 muestras, aumentando así notablemente el tiempo necesario para la preparación de un número suficiente de patrones de calibración, nunca inferior a 10.
Como consecuencia de las desventajas comentadas se procedió a diseñar un nuevo generador (Cámara B) que puede ser observado en la Figura 2. La principal diferencia con respecto a la Cámara A consiste en el mayor volumen del dispositivo(1) donde se genera la atmósfera de cuarzo estándar y en cuya parte superior secolocan los muestreadores. Este diseño de borosilicato de 3 mm de grosor dispone de una tapa del mismo material con 4 orificios simétricos (2) por donde se introducen los tubos que conectan las bombas de muestreo a los selectores de fracción respirable. El cuarzo patrón se deposita en la base de un bol (3) construido con el mismo material que el diseño (1). La atmósfera de cuarzo se generaintroduciendo aire comprimido a ambos lados del bol, a través de dos tubos (4) de un diámetro interno de 1 mm, también de borosilicato.
Figura 2.- Cámara B empleada para la preparación de estándares de calibración: 1) Volumen de vidrio donde se genera la atmósfera de polvo; 2) Orificios por donde se introducen las conexiones de los muestreadores a las bombas; 3) Bol donde se deposita el patrón de cuarzo; 4) Introducción de aire comprimido.

Para la preparación de los estándares de calibración se depositan en torno a100 mg del estándar en el bol y se introduce aire comprimido durante unos 2-3 segundos a una presión de 50 psi. Se deja estabilizar durante aproximadamente 1 minuto con objeto de que los aglomerados de partículas que se hayan podido formar sedimenten y finalmente se ponen en funcionamiento las bombas de muestreo. Empleando tiempos de aspiración entre 5 y 20 segundos se pueden obtener fácilmente patrones de cuarzo que cubran el rango 20-500 µg.
La cámara B permite solucionar los problemas de operatividad encontrados con el anterior diseño (Cámara A), ya que se pueden preparar en cada ronda 4 estándares a la vez y, por otro lado, el mayor volumen del diseño donde se genera la atmósfera de cuarzo y el menor tiempo de inyección de aire comprimido evitan las operaciones de acondicionamiento de polvo en el bol. Por este motivo, la cámara B fue seleccionada para la preparación de los patrones de calibración a lo largo del presente trabajo.
Selección del detector en Difracción de rayos X
El difractómetro utilizado en el presente trabajo dispone de dos detectores: un detector proporcional de Xe y el nuevo detector X´Cellerator basado en la tecnología “Real Time Multiple Strip”, del cual se espera, no sólo una mayor rapidez, sino también un incremento de sensibilidad. A lo largo de esta sección se pretende comparar ambos detectores para la determinación de cuarzo en DRX, especialmente en lo que respecta a los límites de detección y cuantificación obtenidos con cada uno de ellos.
El funcionamiento del difractómetro con cada detector es diferente: en elcaso del proporcional estamos trabajando con un módulo de rendija de recepción programable, mientras que el X´Cellerator se emplea en modo “scanning”. Por tanto, algunas de las condiciones operacionales fueron distintas, tal y como se puede observar en la Tabla 1. La ventaja del X´Cellerator en rapidez de análisis es evidente si se tiene en cuenta el tiempo de barrido empleado en la cuantificación decuarzo sobre su línea principal (26,66º 2Θ). Por otro lado, tal y como veremos acontinuación, la mayor sensibilidad del X´Cellerator permite trabajar con intensidades más bajas (50 mA) lo que a la larga se traduce en una mayor vida del tubo de rayos X.
Tabla 1.- Condiciones operacionales empleadas con el detector proporcional de Xe y el detector X´Cellerator.
Proporc. (Xe) X´Cellerator
Máscara incidente 10 mm 15 mm Óptica de recepción PRS 0,5 mm Active length 2,6 mm Tamaño de paso - 0,02º Tiempo por paso - 50 Velocidad de barrido 0,014º/s 0,05º/s Tiempo de análisis 4´ 1´14´´
La optimización del tamaño y tiempo de paso empleados con el X´Cellerator se llevó a cabo midiendo 10 réplicas de dos membranas de Ag con un depósito teórico de cuarzo de 21 y 499 µg respectivamente, y analizando las precisiones obtenidas. En un principio se seleccionó un tamaño de paso de 0,02º y se varió el tiempo de paso entre 20 y 200 segundos. A continuación se aumentó el tamaño de paso a 0,04º, variando el tiempo entre 50 y 200 segundos. Los resultados se recogen en la Tabla 2.
En general las precisiones obtenidas fueron del mismo orden de magnitud, no existiendo diferencias significativas, por lo que se seleccionó un tamaño de paso de 0,02º con un tiempo de conteo de 50 s como valores de compromiso entre precisión y tiempo de análisis.
Cálculo del límite de detección y cuantificación empleando un detector proporcional de Xe
El límite de detección (LD) se calcula como aquella concentración de cuarzo (µg) que proporciona una señal (y): y= yblanco + 3σblancodonde:yblanco es la señal del blanco, pudiendo tomar como tal la media de 10 medidas de un blancoσblanco es la desviación típica del blanco. Se puede tomar como tal la desviación típica de 10 medidas de un blanco
Tabla 2.- Precisiones obtenidas (%RSD) para 2 membranas de Ag con depósitos teóricos de 21 y 499 µg empleando diferentes condiciones de barrido en el X´Cellerator.
Tiempo por paso (s) %RSD (21µg) %RSD (499 µg)
Tamaño de paso 0,02º20 29 0,850 12 1100 9 0,4200 10 0,3
Tamaño de paso 0,04º50 23 0,3100 13 0,5200 6 0,3
Para el cálculo del límite de detección se calcinaron 10 filtros de PVC en blanco y se prepararon las correspondientes membranas de plata. La media de las señales de los 10 blancos fue yblanco=0,5, con una desviación estándar σblanco =1,3.
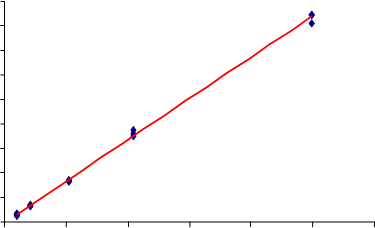
La recta de calibrado obtenida con el detector proporcional se muestra en la Figura 3. Se prepararon 15 patrones de calibración a partir de dos suspensiones de cuarzo NIST 1878a (20,12 y 50,07 mg llevados a 1 L de alcohol) y tomando alícuotas de 1, 2, 10, 20 mL de la 1ª suspensión y 10 mL de la 2ª, después de 1 hora sometida a ultrasonidos. El límite de detección obtenido fue LD = 5 µg.
El límite de cuantificación teórico se calcula como aquella concentración de cuarzo (µg) que proporciona una señal (y): y= yblanco + 10σblancodonde:yblanco es la señal del blanco, pudiendo tomar como tal la media de 10 medidas de un blancoσblanco es la desviación típica del blanco. Se puede tomar como tal la desviación típica de 10 medidas de un blanco
En nuestro caso, teóricamente, LC=16 µg, si bien se puede considerar como límite de cuantificación real 20 µg dado que este nivel de concentración fue uno de los tres niveles ensayados para el cálculo de la exactitud, repetibilidad, reproducibilidad e incertidumbre en la validación del método INS-IT05 “Instrucción de trabajo para la determinación de sílice libre cristalina en materia particulada (fracción respirable) porIR”.
500450400350
![]()
300250200150100500y = 0.8602x - 2.6311 R2= 0.99830 100 200 300 400 500 600

MICROGRAMOS
Cálculo del límite de detección y cuantificación empleando un detector X´Cellerator
Los blancos utilizados para el cálculo de los límites de detección y cuantificación, así como los patrones de calibración empleados para construir la recta de calibrado (Figura 4) fueron exactamente los mismos que los utilizados en el apartado anterior. De esta manera se redujo al mínimo la influencia de otros factores tales como la preparación de muestras sobre el resultado final, de forma que la diferencia en sensibilidad obtenida fue exclusivamente debida a la utilización de un determinado tipo de detector.
Se midieron 10 blancos obteniendo una señal promedio yblanco=0,4 con una desviación típica σblanco = 0,4. A partir de estos valores y la pendiente de la recta de calibrado se calculó un LD de 3 µg y un LC de 7 µg, aproximadamente la mitad delos obtenidos con el detector proporcional.
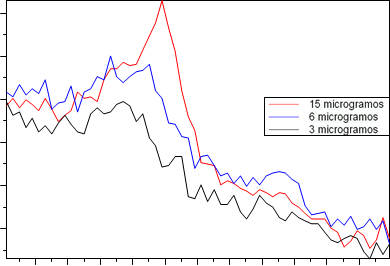
El X´Cellerator reduce el error asociado a la estadística de conteo, gracias a su diseño, que posibilita un incremento de sensibilidad a la hora de cuantificar muy bajas concentraciones de cuarzo (los cristalitos no perfectamente paralelos al plano de la muestra contribuyen también a la difracción) y un aumento de la velocidad de medida respecto a los detectores convencionales. Así, en la Figura 5 podemos observar la capacidad del X´Cellerator para detectar 3, 6 y 15 µg de cuarzo respirable sobre membranas de PVC.
180
160
140
120
100
![]()
80
60
40
20
0
y = 0.337x - 0.5205 R2 = 0.9986
0 100 200 300 400 500 600
MICROGRAMOS
Figura 5.- Difractogramas obtenidos para 3, 6 y 15 µg de cuarzo sobre filtros de PVC empleando un detector X´Cellerator.
340
 |
|
330320310300290 |
26.40 26.50 26.60 26.70 26.80 26.90 27.00 27.10 27.20 27.30 27.402Theta (°)
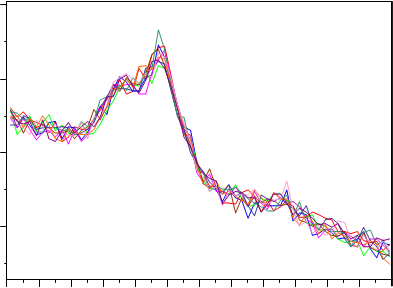
Además de la sensibilidad, la precisión es sumamente importante en un análisis cuantitativo. En DRX esta precisión depende generalmente del tiempo necesario para medir la muestra, de tal manera que si queremos aumentar por dos la precisión deberíamos multiplicar por cuatro el tiempo de análisis. En la Figura 6 se muestran los difractogramas obtenidos para 10 réplicas de una membrana de PVC con un depósito teórico de 10 µg de cuarzo. La precisión obtenida (%RSD) resultó ser del 15% lo que confirma que el X´Cellerator no sólo proporciona datos más rápido sino también más precisos, precisamente por su capacidad para acumular cuentas en cada punto durante muchos segundos en este tipo de barridos cortos.
Figura 6.- Difractogramas obtenidos para 10 réplicas de un filtro de PVC con un depósito teórico de 10µg.
360
 |
|
340320300 |
26.30 26.40 26.50 26.60 26.70 26.80 26.90 27.00 27.10 27.20 27.30 27.40 27.502Theta (°)
Selección del tipo de filtro a emplear en análisis directo de sílice sobre membrana
La I.T.C. 2.0.02 establece dos parámetros de control para preservar la salud de los trabajadores: la concentración de la fracción respirable y la concentración de sílice libre cristalina existentes en la atmósfera de trabajo, ambas en mg/m3. El muestreo de la fracción respirable del polvo se realiza mediante filtración utilizando un selector de tipo ciclónico capaz de separar la fracción respirable del polvo total existente en el ambiente.
En España, desde el año 2001, las muestras procedentes de minería de exterior han sido tomadas sobre filtros de PVC debido a las mejores prestaciones analíticas que ofrecen en comparación con los filtros de celulosa utilizados hasta entonces por el INS, tanto para la determinación gravimétrica de la fracción respirable como para la determinación de sílice cristalina en dicha fracción [6].
El empleo de un determinado tipo de filtro juega un papel fundamental en los métodos directos de análisis, ya que ahora es el propio filtro el soporte introducido en el instrumento de medida y, por tanto, debemos escoger el filtro más adecuado para la determinación de sílice tanto por IR como por DRX. Ahora bien, dado que otro parámetro de riesgo a determinar es la concentración de fracción respirable debemos escoger un tipo de filtro de compromiso entre las dos determinaciones, gravimetría y sílice, y entre las dos técnicas empleadas, puesto que en la mayor parte de los casos la muestra es tomada por un higienista y el laboratorio no conoce hasta el momento de la recepción del filtro la matriz en la que se encuentra el cuarzo y, por consiguiente, la técnica más adecuada para su determinación.
Una diferencia de los métodos directos de análisis con respecto a los indirectos es que en los primeros se trabaja con filtros de 25 mm y no de 37 mm de diámetro, para conseguir que la mayor parte de la muestra sea expuesta a la
Espectroscopía de IR.
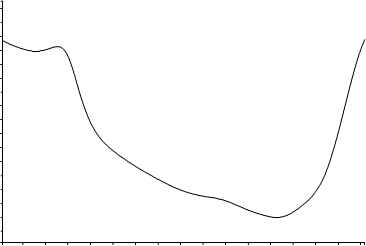
La determinación directa de SiO2 mediante IR se lleva a cabo sobre el espectro diferencia obtenido entre el espectro del filtro con muestra y el espectro del filtro “en blanco”, es decir, sin muestra tomada, El cuarzo presenta sus bandas de interés en la zona 820-750 cm-1, por lo que es sumamente importante que el filtro utilizado no presente bandas de absorción en dicha zona. Tal y como se observa en la Figura 7 un filtro de PVC presenta una línea plana en ese intervalo lo que permite el empleo de este tipo de filtros para el análisis directo sobre membrana.
Existen otro tipo de filtros de copolímeros PVC-acrilonitrilo que presentan espectros IR similares sin bandas de absorción en la zona analítica de interés, sin embargo se ha desechado su empleo para este trabajo porque este tipo de membranas tienen tendencia a deflagrar durante el proceso de calcinación en la mufla proporcionando malas recuperaciones analíticas tal y como se puede observar en la Tabla 3 donde se recogen los resultados obtenidos en la determinación de cuarzo sobre 10 filtros de este tipo con un depósito teórico de100 µg. Por tanto, no sería posible llevar a cabo una comparación “real” entremétodos directos o indirectos.
Los filtros de Ag, ampliamente utilizados en DRX, quedan descartados para el análisis directo en IR porque la absorción de radiación es tal que satura el detector.
Difracción de rayos X.
El empleo de filtros de Ag como soporte de medida en DRX empleando métodos de redeposición es muy habitual [7,8]. Proporcionan muy buenas relaciones señal/ruido y además permiten utilizar la propia señal de Ag para corregir posibles efectos de absorción de radiación o de profundidad del depósito de muestra. Sin embargo, la utilización de este tipo de filtros en métodos directos de análisis no lo es tanto, dado que por el propio material del filtro y su porosidad (0,45 µm), ofrecen una gran resistencia al paso de aire a su través, lo que ocasiona problemas en el funcionamiento de las bombas durante las 8 horas que suele durar la toma de muestra.
Empleando la cámara B comentada en apartados anteriores se procedió a preparar patrones de calibración en el rango 20-500 µg sobre membranas de Ag y PVC, empleando como muestreador un selector IOM. La recta de calibrado obtenida con membranas de Ag tenía una pendiente de calibración de aproximadamente el doble que la obtenida con PVC (Figura 8), lo que puede considerarse una medida de la sensibilidad.
0,33
 |
|
0,310,29 |
0,27
 |
|
0,250,230,210,190,17 |
850 830 810 790 770 750 730 710
Número de onda (cm-1)
Tabla 3.- Resultados obtenidos en la determinación de SiO2 empleando membranas de copolímero PVC- acrilonitrilo.
|
Membrana |
Depósito teórico SiO2 (µg) |
Valor obtenido (µg) |
% RSD (n = 10) |
|
DM-450 |
100 ± 1 |
43 ± 22 |
50 |
A pesar de la mejor sensibilidad obtenida con los filtros de Ag, durante la preparación de los patrones de calibración se observó que las bombas en ocasiones tenían dificultades para mantener el caudal de aspiración. De hecho, esta preparación sólo pudo ser llevada a cabo empleando bombas APEX (Casella), de última generación, mientras que bombas más antiguas como las AFC 123 (Casella) fueron incapaces de mantener el caudal de aspiración y se paraban. Estos problemas relacionados con la toma de muestra, unidos al mayor coste de las membranas de Ag en comparación con las de PVC (aproximadamente el triple), obligó a escoger estas últimas como las más adecuadas para llevar a cabo el análisis directo sobre membrana.
Figura 8.- Rectas de calibrado obtenidas en la determinación directa de cuarzo mediante DRX sobre membranas de Ag y PVC.
300
|
PVC Ag |
250
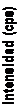
200
150
100
50
0
y = 0.5876x - 5.4444 R2 = 0.9983
y = 0.3104x + 1.2553 R2 = 0.9987
0 100 200 300 400 500
Microgramos de cuarzo
Comparación de los métodos directos (IR y DRX) e indirectos (IR) para la determinación de cuarzo α en muestras de polvo respirable
La comparación de los dos tipos de metodologías se llevó a cabo empleando muestras de cuarzo α NIST 1878a, preparadas en la cámara B comentada anteriormente, de la misma forma que la indicada para los patrones de calibración, y empleando filtros de PVC de 25 mm por las razones explicadas en la sección anterior. Asimismo, se compararon dos tipos de selectores de fracción respirable: IOM (SKC) a un caudal de 2 l/min y Higgins Dewell (Casella) a un caudal de aspiración de 2,2 l/min. Ambos muestreadores llevan acabo una separación de la fracción respirable según el criterio definido en la norma UNE-EN 481 [9].
El depósito teórico de cuarzo, tanto en los patrones de calibración como en las muestras a analizar, se calculó mediante diferencia de pesada de los filtros, antes y después de la toma de muestra, empleando una balanza analítica de sensibilidad 0,001 mg para poder pesar a nivel de µg. La incertidumbre asociada a la determinación gravimétrica del contenido en cuarzo sobre los filtros de PVC resultó ser de 2 µg para un factor de cobertura de k=2 (al 95% del intervalo de confianza).
En total se prepararon 73 membranas de PVC con depósitos de cuarzo patrón empleando un muestreador IOM, y 73 membranas empleando un ciclón Higgins Dewell. En la Tabla 4 se recogen la media y los valores máximos y mínimos del contenido en cuarzo de las membranas preparadas con cada muestreador.
Tabla 4.- Depósitos teóricos de cuarzo contenidos sobre filtros de PVC obtenidos con un ciclón Higgins Dewell y un selector IOM.
|
Muestreador |
Número de |
Media (µg) |
Desviación |
Mínimo |
Máximo |
|
muestras |
estándar |
(µg) |
(µg) |
||
|
IOM |
73 |
287 |
270 |
8 |
1287 |
|
Higgins Dewell |
73 |
158 |
138 |
15 |
722 |
Las 146 muestras de cuarzo fueron analizadas directamente sobre el propio filtro de PVC, tanto mediante FTIR como mediante DRX, y posteriormente se sometieron a un proceso de calcinación para finalmente determinar el contenido en cuarzo mediante FTIR, siguiendo el procedimiento INS-IT05 “Instrucción de trabajo para la determinación de sílice libre cristalina en materia particulada (fracción respirable) por IR”. El motivo de escoger este método analítico, y no el basado en DRX, se debió a que el tiempo de análisis es mucho menor, tanto por la preparación de muestras como por la propia instrumentación, y a que proporciona prestaciones analíticas similares. En definitiva, se trata de comparar dos metodologías basadas en el análisis directo sobre membrana con respecto a un método indirecto perfectamente validado.
Como hemos comentado con anterioridad, en los métodos directos de análisis es el propio filtro el que se emplea como soporte para la medición de la muestra, en lugar de la pastilla de KBr o la membrana de Ag sobre la que se redepositaba el residuo de calcinación en los métodos indirectos. Por tanto, lo único que debemos seleccionar son los parámetros instrumentales a emplear en el espectrofotómetro FTIR o el difractómetro de rayos x. Estas condiciones operacionales se indican en las Tablas 5 y 6.
Las Figuras 9 y 10 muestran una distribución del contenido en cuarzo de los filtros en forma de histograma.
Los resultados obtenidos en la determinación directa de cuarzo sobre membrana de PVC mediante FTIR (IR DOF) y DRX (DRX DOF) se representan en las Figuras 11-14 frente a los resultados obtenidos en la determinación indirecta de cuarzo por FTIR (IR KBr), para los dos tipos de muestreadores empleados, el ciclón Higgins Dewell y el selector IOM. Lógicamente, por ser el método indirecto el “verdadero” se representó en el eje de abscisas. La representación se ha realizado para contenidos de cuarzo entre 3 y 500 µg, por ser éste el rango en el que el método indirecto está validado.
En la Tabla 7 se recogen los coeficientes de las rectas de regresión obtenidas, así como los intervalos de confianza para la pendiente. Ninguna de las ordenadas en el origen resultó ser significativa frente a cero (P > 0,05).
Figura 9.- Distribución por frecuencias del contenido en cuarzo sobre membranas de PVC preparadas con un selector IOM.
IOM
20
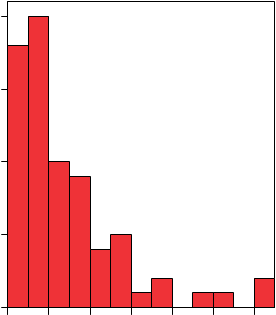 |
|
15 |
10
 |
|
50 |
0,00 200,00 400,00 600,00 800,00 1000,00 1200,00
Figura 10.- Distribución por frecuencias del contenido en cuarzo sobre membranas de PVC preparadas con un ciclón Higgins Dewell.
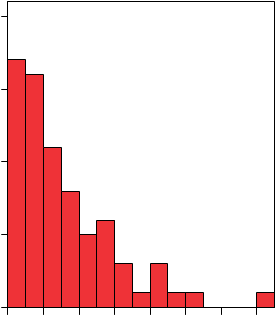
Higgins Dewell
20
 |
|
15 |
10
|
|
50 |
0,00 100,00 200,00 300,00 400,00 500,00 600,00 700,00
Tabla 5.- Condiciones instrumentales empleadas en el espectrofotómetro FTIR.
Instrumento Spectrum One
Rango de análisis 1000-370 cm-1 Acumulaciones 16
Detector MIR TGS
Fuente MIR
Divisor de haz OptKBr
Resolución 4,00 cm-1
Apodización Fuerte
Tipo de haz Ratio
Velocidad OPD 0,20 cm/s
Tamaño de J-Stop 8,94 mm
Tipo de interferograma Doble Dirección de barrido Combinada
Tipo de IR FT
Tabla 6.- Condiciones instrumentales empleadas en el difractómetro de rayos X.
Instrumento Difractómetro PW3040/60 Tubo 2.2 kW LFF Cu
Potencia 40 kV / 50 mA
Rendija Soller 0.04 rad
Haz incidente
Rendija de divergencia 1º
Máscara fija 15 mm
Rendija Soller 0.04 rad
Haz difractado
Módulo X´Cellerator
Filtro Ni
Sample Spinner 1 Rev/s
Cuarzo (3.343 Å) 26.40 to 27.20 º2Θ
Tamaño de paso 0,02º 2Θ
Tiempo por paso 50 s
Velocidad de barrido 0,05º 2Θ/s
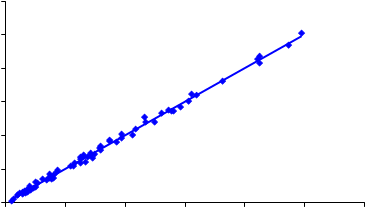
600

500
 |
|
y = 1.0059x + 1.2276 R2 = 0.99594003002001000 |
0 100 200 300 400 500 600
IR KBr (µg de cuarzo)
Figura 12.- Relación entre el contenido en cuarzo medido por IR (método indirecto, KBr) y el resultado obtenido mediante IR (método directo), empleando como muestreador un ciclón Higgins Dewell.
600
500
 |
|
y = 1.0014x + 2.0579 R2 = 0.99674003002001000 |
0 100 200 300 400 500 600
IR KBr (µg de cuarzo)
600
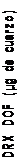
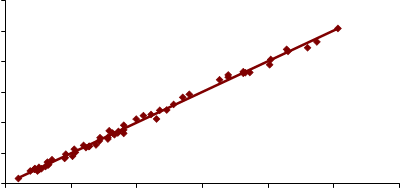
500
y = 1.0061x + 0.3769 R2 = 0.9961
400
300
200
100
00 100 200 300 400 500 600
IR DOF (µg de cuarzo)
Figura 14.- Relación entre el contenido en cuarzo medido por IR (método indirecto, KBr) y el resultado obtenido mediante IR (método directo), empleando como muestreador un selector IOM.
600
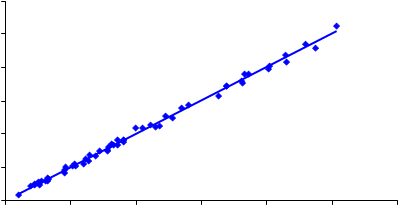
500
 |
|
y = 1.0022x + 1.5986 R2 = 0.99674003002001000 |
0 100 200 300 400 500 600
IR KBr (µg de cuarzo)
Método Y = a*X + b IC (a)
a b
Ciclón Higgins Dewell
|
IR DOF |
1,001 |
2,050 |
0,988 – 1,015 |
|
DRX DOF |
1,006 |
1,268 |
0,99 – 1,021 |
|
Selector IOM |
|||
|
IR DOF |
1,002 |
1,594 |
0,987 – 1,018 |
|
DRX DOF |
1,006 |
0,438 |
0,989 – 1,022 |
(KBr)
Y = contenido en cuarzo obtenido mediante método directo de análisis
X = contenido en cuarzo obtenido mediante método indirecto de análisis IC = Intervalos de confianza para la pendiente (95%)
Tal y como se observa en la Tabla 7 las pendientes de las rectas de regresión obtenidas no difieren significativamente de la unidad en ninguno de los casos, por lo que podemos decir que no existen diferencias sistemáticas entre el empleo del método indirecto de análisis por IR y los métodos directos (IR y DRX). Por otra parte, si se comparan los métodos directos entre sí (IR y DRX) las pendientes de las rectas de regresión obtenidas para el empleo del ciclón Higgins Dewell y el selector IOM tampoco difieren significativamente de la unidad (ver Tabla 8), lo que en principio excluye cualquier tipo de diferencia entre ambos.
Tabla 8.- Coeficientes obtenidos para las rectas de regresión correspondientes a la comparación entre métodos directos de análisis.
Muestreador Y = a*X + b IC (a)
a b
Higgins Dewell 1,000 0,579 0,985 – 1,015
IOM 1,003 -1,074 0,983 – 1,022
Y = contenido en cuarzo obtenido mediante método directo de análisis(DRX)
X = contenido en cuarzo obtenido mediante método directo de análisis (IR) IC = Intervalos de confianza para la pendiente (95%)
600
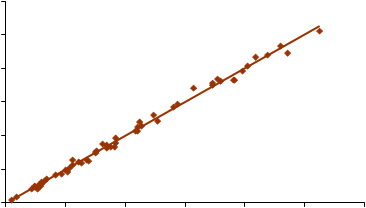
500
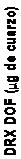 |
|
y = 1.0029x - 1.1504 R2 = 0.99494003002001000 |
0 100 200 300 400 500 600
IR DOF (µg de cuarzo)
Figura 16.- Relación entre el contenido en cuarzo medido por IR (método directo) y el resultado obtenido mediante DRX (método directo), empleando como muestreador un ciclón Higgins Dewell.
600
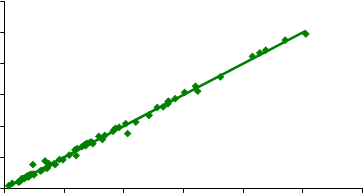
500
 |
|
y = 0.9999x + 0.5078 R2 = 0.99624003002001000 |
0 100 200 300 400 500 600
IR DOF (µg de cuarzo)
Validación y cálculo de la incertidumbre global para la determinación de cuarzo α mediante métodos directos de análisis
La cámara B empleada para la preparación de muestras (membranas de PVC con depósitos de cuarzo) no permite obtener réplicas de un determinado nivel de concentración por lo que la validación no se pudo llevar a cabo de la misma forma que con los métodos indirectos de análisis: preparar membranas a 3 niveles de concentración y analizarlas durante días sucesivos. Por esta razón, la validación se realizó a partir del estudio de las recuperaciones obtenidas en el rango analítico de interés: desde el límite de cuantificación hasta 500 µg de cuarzo. La recuperación se calculó a partir de la siguiente expresión:
donde:
Recuperación (%) = X
VR
×100
X es la concentración de cuarzo obtenida
VR es la concentración de cuarzo real
Exactitud y precisión
Un aspecto importante es determinar si los resultados obtenidos difieren significativamente del valor de referencia (100 %). Para ello se calculó el índice de compatibilidad de acuerdo a la siguiente expresión:
IC =
(UVM
V − X
≤ 2
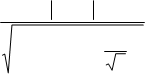 |
|
2 |
n
donde:
V es el valor de referencia (100 %)
X es la recuperación media obtenida con cada método (IR o DRX) y muestreador (Higgins Dewell o IOM)
UVM
es la incertidumbre combinada del valor de referencia, que a su vez
viene dada por la expresión
UVM =
| U |
| + U |
2 2
MR GR
![]()
, donde
U MR es la incertidumbreasociada al propio material de referencia obtenida del certificado de calibración delcuarzo NIST 1878a (0,105), y U GRes la incertidumbre asociada a la determinacióngravimétrica del contenido de cuarzo sobre membranas de PVC (0,002 mg sobre un filtro de PVC de 6 mg dan una incertidumbre relativa del 0,033 %)
S es la desviación típica de las valores medios de recuperaciónn es el número de muestras analizadas
W es el factor Wecc 19
Tabla 9.- Estadísticos descriptivos para las recuperaciones obtenidas en la determinación directa de cuarzo α mediante IR y DRX empleando dos tipos de muestreadores.
|
Método |
Media |
Intervalo de confianza al 95% |
Mediana |
Mínimo |
Máximo |
Desviación típica |
|
Ciclón Higgins Dewell |
||||||
|
DRX |
100,9 |
99,7 – 102,1 |
101,4 |
88,8 |
111,6 |
4,8 |
|
IR |
101,1 |
99,9 – 102,3 |
102,1 |
79,1 |
109,5 |
4,8 |
|
Selector IOM |
||||||
|
DRX |
99,8 |
98,7 – 101,0 |
100,2 |
84,1 |
108,9 |
4,8 |
|
IR |
100,8 |
99,7 – 101,9 |
101,3 |
85,2 |
110,7 |
4,3 |
Figura 17.- Recuperaciones (%) obtenidas para la determinación directa de cuarzo α mediante DRX e IR empleando un ciclón Higgins Dewell (DRXHD e IRHD) y un selector IOM (IRIOM y DRXIOM).
120

 |
|
120 |
110110
100100
90 90
80
120

IRIOM

80
120

DRXIOM

110110
100100
90 90
80
IRHD80
DRXHD
Para los cuatro casos analizados el índice de compatibilidad resultó ser inferior a 2, es decir, los métodos analíticos propuestos son exactos, proporcionan resultados que no difieren significativamente del valor verdadero. La Tabla 10 muestra un resumen de las exactitudes y precisiones obtenidas con cada método y muestreador utilizado.
Límites de detección y cuantificación
Ya habíamos indicado con anterioridad la forma en que se definen y calculan los límites de detección y cuantificación, y su aplicación para la comparación de dos detectores en DRX, en el caso de métodos indirectos de análisis, donde los patrones de calibración se preparaban por filtración a partir de una suspensión del patrón de cuarzo en alcohol. Por tanto, cuando se llevan a cabo métodos directos de análisis los límites de detección y cuantificación serán distintos ya que los patrones se preparan empleando muestreadores de polvo respirable en cámaras donde se genera una atmósfera de cuarzo con esa distribución granulométrica. En el caso dela DRX la distribución del patrón sobre los filtros de PVC difiere en homogeneidad con relación a la obtenida en los procedimientos de filtración empleados en los métodos de redeposición. Esta diferencia es aún mayor en el IR, ya que en los métodos indirectos se emplean pastillas de KBr de 13 mm de diámetro con lo que el patrón está más concentrado en el área circular de aproximadamente 7 mm que excita la radiación IR en comparación con la distribución sobre un filtro de PVC de 25 mm.
Tabla 10.- Exactitudes y precisiones obtenidas en la determinación de cuarzo α mediante métodos directos de análisis.
Método Exactitud (*)
Precisión (*)
V − X
V X
Ciclón Higgins Dewell
DRX 0,9 4,8
IR 1,1 4,7
Selector IOM
DRX 0,2 4,8
IR 0,8 4,3
(*) V es el valor “verdadero”
X es la recuperación media obtenida con cada método (IR o DRX) y muestreador (Higgins Dewell o IOM)
S es la desviación típica de las valores medios de recuperación
El distinto funcionamiento de los muestreadores empleados en este proyecto provocó una distribución diferente de la muestra sobre el filtro, más homogénea en el IOM y más concentrada en el centro del filtro en el caso del ciclón Higgins Dewell. Así, cuando se calculó la precisión, expresada como desviación estándar relativa, para 8 medidas de un mismo filtro rotando el mismo aproximadamente 25º en cada barrido, resultó ser del 0,5% con el selector IOM, mientras que para el ciclón Higgins Dewell se situó en el 1%. Por ello, se encontraron ligeras diferencias en la sensibilidad obtenida con cada muestreador, especialmente en el caso del IR, si bien en ambos casos fueron del mismo orden de magnitud. Por sensibilidad entendemos el gradiente de la curva señal - concentración, es decir, la pendiente de la recta de calibrado. La Tabla 11 recoge los límites de detección y cuantificación así como las sensibilidades obtenidas a partir de las rectas de calibrado y utilizando como blancos filtros sin usar del mismo lote que los empleados en la preparación de muestras.
Tabla 11.- Sensibilidad, límites de detección (LD) y límites de cuantificación (LC) obtenidos en la determinación directa de cuarzo α empleando diferentes muestreadores.
Método Sensibilidad LD (µg) LC (µg)
Ciclón Higgins Dewell
|
IR |
0,0007 Abs/µg |
2 |
7 |
|
DRX |
0,423 cps/µg |
3 |
10 |
|
Selector IOM |
|||
|
IR |
0,0005 Abs/µg |
3 |
10 |
|
DRX |
0,366 cps/µg |
3 |
10 |
Cálculo de la incertidumbre
Los test de recuperaciones, como los efectuados en este trabajo, permiten estimar el error sistemático o sesgo. En este sentido, los resultados obtenidos pueden servirnos para calcular la incertidumbre combinada de los métodos directos de análisis a partir de la siguiente fórmula:
2 2
U sesgo =
|
donde: RMSsesgo + UVM |
UVM
es la incertidumbre combinada del valor de referencia, que habíamosvisto con anterioridad (0,11)
RMS
sesgo =
Σ(sesgo)2
n
![]()
, donde el sesgo es la diferencia entre larecuperación obtenida para cada muestra y el valor de referencia aceptado (100%), y n es el número de muestras analizadas
La Tabla 12 recoge las incertidumbres combinadas obtenidas con los métodos directos de análisis (IR y DRX) y con el método indirecto (IR) basado en la preparación de pastillas de KBr.
En el caso de los métodos directos de análisis las incertidumbres obtenidas se situaron por debajo del 5%, mientras que en la determinación de cuarzo mediante IR empleando el método indirecto del KBr fueron ligeramente superiores, en torno al 6%, lo que podría explicarse debido a la manipulación que conlleva la preparación de la muestra y los riesgos de pérdidas del analito que ello supone.
Tabla 12.- Incertidumbres combinadas obtenidas en la determinación de cuarzo α mediante métodos directos de análisis (IRDOF y DRXDOF) e indirectos (IRKBr).
Método Incertidumbre combinada (%)
Ciclón Higgins Dewell
DRXDOF 4,9
IRDOF 4,9
IRKBr 6,2
Selector IOM
DRXDOF 4,7
IRDOF 4,4
IRKBr 6,0
CONCLUSIONES
a) Se ha desarrollado una cámara de vidrio para la generación de atmósferas de polvo respirable que permite la preparación de patrones de calibración para la determinación directa de cuarzo α en el rango entre 10 y 1000µg, relativamente rápido, empleando menos de 1 minuto en la preparación simultánea de cuatro estándares.
b) De la comparación de diferentes detectores en DRX para la determinación directa de cuarzo α, el X´Cellerator, basado en la tecnología “Real Time Multiple Strip”, proporciona una mejor sensibilidad, lo que conduce a límites de cuantificación de aproximadamente la mitad que los obtenidos con un detector proporcional, con la ventaja que ello supone para la determinación de cuarzo a niveles de los actuales valores límite, especialmente si es preciso recurrir a líneas de cuarzo menos sensibles.
c) El PVC resultó ser el tipo de filtro más adecuado para el análisis directo de muestras de polvo respirable, frente a otros como la Ag que no podían ser empleados en IR y además presentaban problemas para el mantenimiento del caudal de aspiración de las bombas. En todo caso el diámetro del filtro debe ser de25 mm con objeto de asegurar que la mayor parte del depósito de muestra seaexpuesto a la radiación IR o a los rayos x.
4) Los dos tipos de muestreadores utilizados en la preparación de muestras, el ciclón Higgins Dewell y el selector IOM, proporcionaron resultados similares en el análisis directo de aquellas, a pesar de que la deposición sobre el filtro es diferente, más uniforme en el IOM frente a la más concentrada en la parte central del filtro en el Higgins Dewell.
5) Fue posible el desarrollo de dos metodologías analíticas basadas en la determinación directa de cuarzo α mediante DRX y FTIR, sin que se observasen diferencias significativas con respecto a un método de redeposición por IR, perfectamente validado. En todo caso, las incertidumbres obtenidas con el empleo de los métodos directos fueron un 20% mejores, en términos relativos, que las obtenidas con el método indirecto, debido a la ausencia de etapas de preparación previas al análisis de la muestra.
REFERENCIAS
- 1. SRI International, Menlo Park, CA, (1983) p. 1156
- 2. Pickard, K. J., Walker, R. F., West, N. G. (1985) Ann. Occup. Hyg., 29, 149
- 3. Lorberau, C. D., Carsey, T. P., Fischbach, T. J. (1990) Appl. Occup. Environ. Hyg., 5, 27
- 4. Addison, J. IOM report TM/91/10 p. 149
- 5. Stacey, P., Tylee, B., Bard, D. (2003) Ann. Occup. Hyg., 47, 269
- 6. Fernández Rodríguez, P., Tesis doctoral, Universidad de Oviedo (2006)
- 7. NIOSH Manual of Analytical Methods (NMAM), Fourth Edition: “Silica, Crystalline, by XRD”, Method 7500
- 8. Método del Laboratorio del Departamento técnico del INS: INSIT10 “Instrucción de trabajo para la determinación del contenido en cuarzo alfa en materia particulada (fracción respirable) por DRX”
- 9. CEN/TR 15230 “Workplace atmospheres Guidance for sampling of inhalable, thoracic and respirable aerosol fractions”, Agosto 2005
Papers relacionados











